Scientific Design
I conducted this work while I was a PhD candidate in the Durocher Lab, part of the Lunenfeld-Tanenbaum Research Institute affiliated with the Department of Molecular Genetics at the University of Toronto. My lab used cutting-edge functional genomics, such as CRISPR/Cas9 whole genome screens to interrogate the DNA damage response and genome stability factors. I had a broad interest in DNA repair and cell cycle control, especially the response to DNA double-strand breaks. From my experience as a PhD researcher, I honed knowledge translation skills in information design and illustration, working with industry-standard tools like Adobe Creative Suite, R, Python, and others.
Purpose
PhD Research
Type
Information Design
Timeline
A while :)
Design Process
Functional genomics
Using CRISPR/Cas9 screens to elucidate the mechanism of action of small molecule inhibitors
In my PhD thesis, I used Toronto Knock-Out (TKO) whole-genome guide RNA libraries developed by Jason Moffat and Traver Hart to study two classes of enzyme inhibitors. CRISPR/Cas9 can be targeted to genomic loci by a unique 20 nucleotide sequence known as the guide RNA. At the target site, Cas9 induces a double-strand break which can be repaired by error-prone mechanisms to produce small insertions or deletions. These mutations compromise the expression of the targeted gene, resulting in highly penetrant loss-of-function gene editing. When applied in pooled format, we can systematically interrogate genetic relationships between the protein-coding genome and any given perturbation (e.g. a single loss-of-function gene or a chemical inhibitor). In the context of chemical screens, we can recover both sensitizing (negative selection; loss of the gene sensitizes to the drug) and suppressor (positive selection; loss of the gene is associated with drug resistance) genes. For my studies I used TP53-deficient hTERT immortalized RPE1 retinal pigmented epithelial cells to recover gene depletions that would otherwise be lost to TP53-mediated cell cycle arrest or cell death, as previously described by my lab.
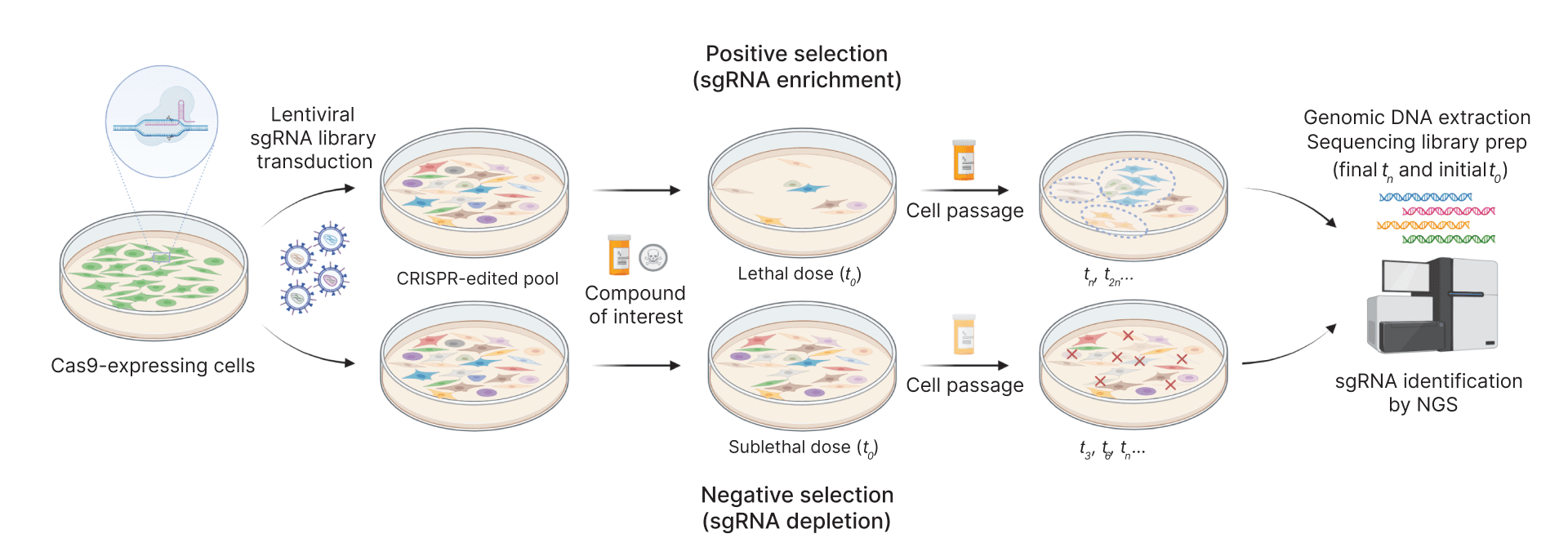
Design of CRISPR/Cas9 whole-genome single-guide RNA library drug screens
Genetic relationships with WEE1 inhibition by adavosertib (AZD-1775)
In this chapter of my PhD thesis, I studied genetic relationships with the clinical WEE1 kinase inhibitor adavosertib (AZD-1775). Prior to this work, no comprehensive genome-wide study had interrogated the consequences of adavosertib treatment in human cells. Unexpectedly, I found robust sensitizing relationships with loss of ribosome quality control (RQC) and suppression of adavosertib toxicity by loss of the eIF2alpha kinase GCN2. I found that adavosertib induces a GCN2-dependent integrated stress response that underlies sensitivity to WEE1 inhibition.
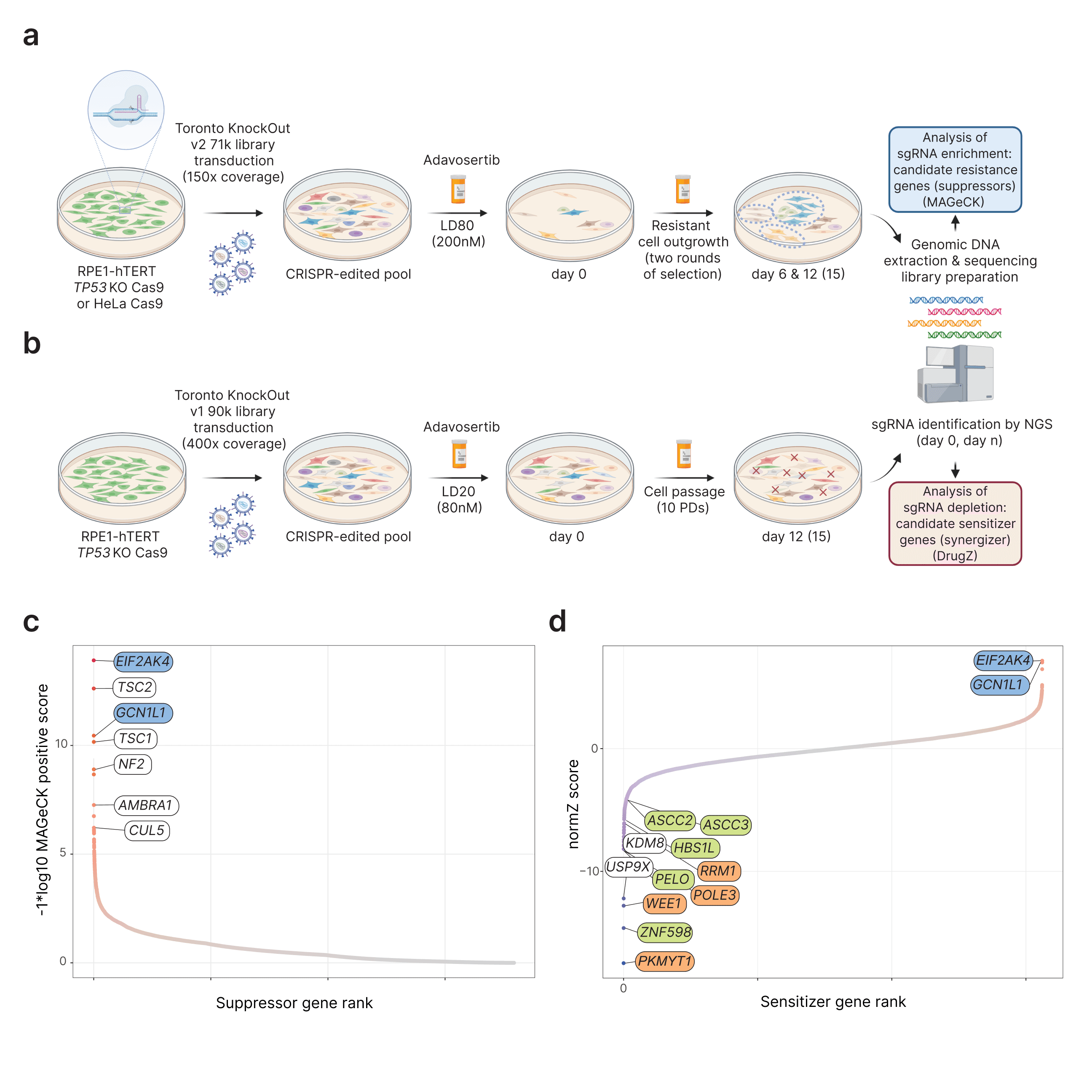
Schematic of selection screens and methodology. Bottom: Gene ranked plots of positive (left) and negative (right) adavosertib screens analyzed by MAGeCK and drugZ respectively. Negative normZ scores indicates sensitization and positive (-LOG10 transformed) MAGeCK scores indicate suppression.
Genetic relationships with MRE11 inhibition by mirin and PFM derivatives
In this chapter of my PhD thesis, I characterize three described MRE11 nuclease inhibitors. Specific MRE11 activities are a key commitment mechanism to direct double-strand breaks (DSB) to faithful repair by homologous recombination (HR). Prior to HR, DSBs must be resected; the process of generating long stretches (likely up to 1kb in cells) 3' single-stranded DNA. In the two-step short and long-range model of end resection published in literature, MRE11 endonuclease activity creates a nick adjacent the DSB. This nick is subsequently used as an entry point for 3'-5' MRE11 exonuclease activity, revealing a small stretch of single-stranded DNA that is required for subsequent enzymes to drive long-range end resection. Although biochemical evidence suggest that these inhibitors inhibit MRE11 in vitro, I leverage TKO libraries to provide comprehensive chemical genetic profiles of mirin, PFM01, and PFM39. Strikingly, I found a strong correlation between PFM01 sensitivity and both CRISPR-mediated loss of mitochondrial gene expression or pharmacological inhibition of mitochondrial translation. Collectively, my work cautions against off-target effects of MRE11 inhibitors when interpreting the phenotypes of their application.
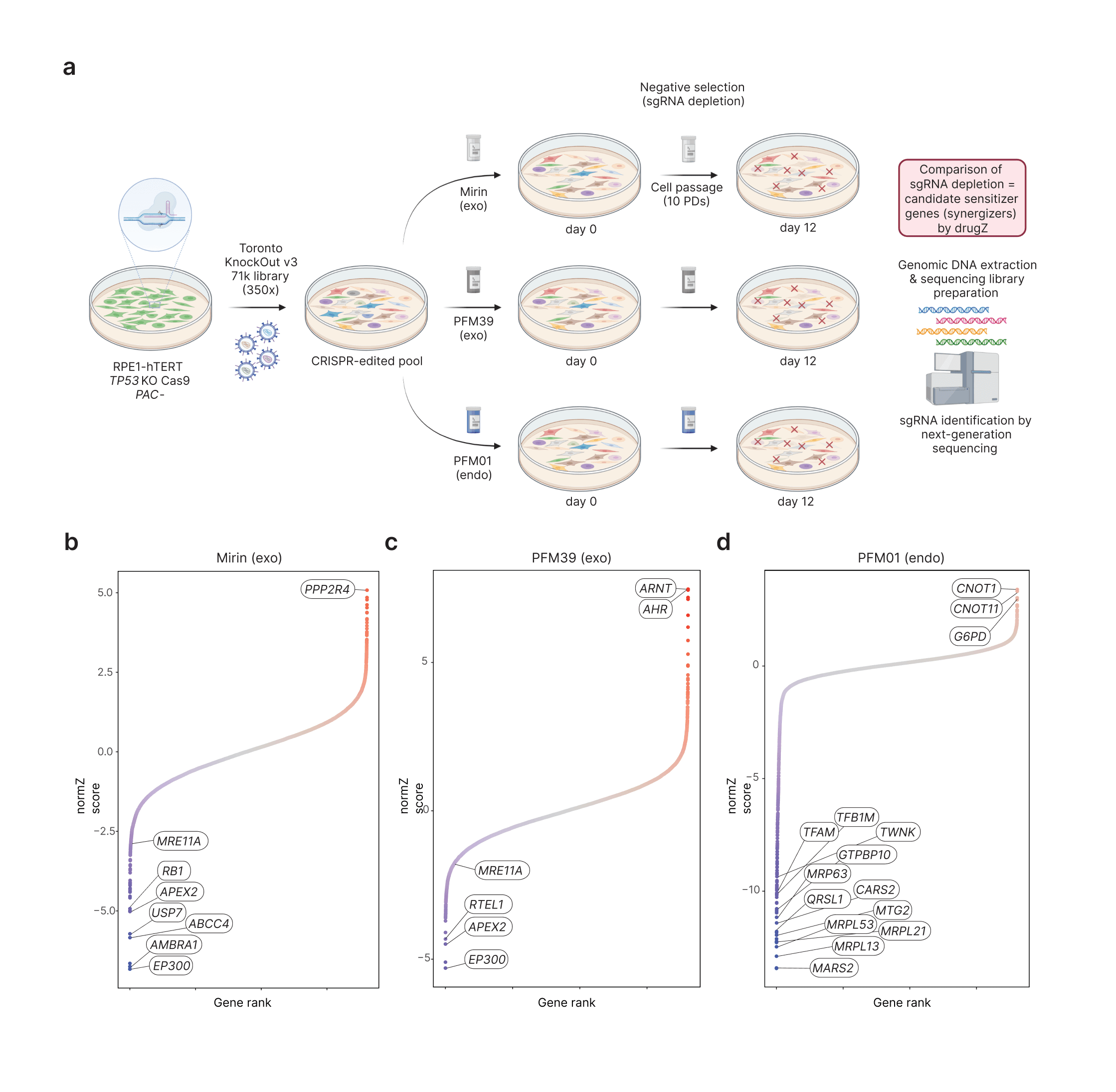
Schematic of MRE11 inhibitor drug screens. Gene ranked plots of drugZ analyzed screens where negative normZ scores indicate sensitization.
DNA double-strand break repair
Control of DNA end resection in mitosis
Repair of DNA double-strand breaks (DSB) is inhibited when cells commit to mitosis to maintain genome stability. This is because mitotic DSB repair is a greater threat than any consequences of delayed repair in the subsequent cell cycle. However, a limited DSB response depending on MDC1 recruitment to DSBs is persistent in mitotic cells. Despite this, I found that DNA end resection, a critical commitment mechanism necessary for HR, is not active at mitotic DSBs. I show that complementation of cells with functional CtIP protein can rescue resection as measured by RPA2 focus formation, a marker for single-stranded DNA. Moreover, unscheduled resection in mitosis is accompanied with a greater burden of DNA lesions marked by the DSB binding protein 53BP1 in the subsequent cell cycle. However, this phenotype is associated with suppression of mitotic cell sensitivity to X-ray ionizing radiation. Taken together, I hypothesize that unscheduled resection channels ends towards alternative end joining mediated repair.
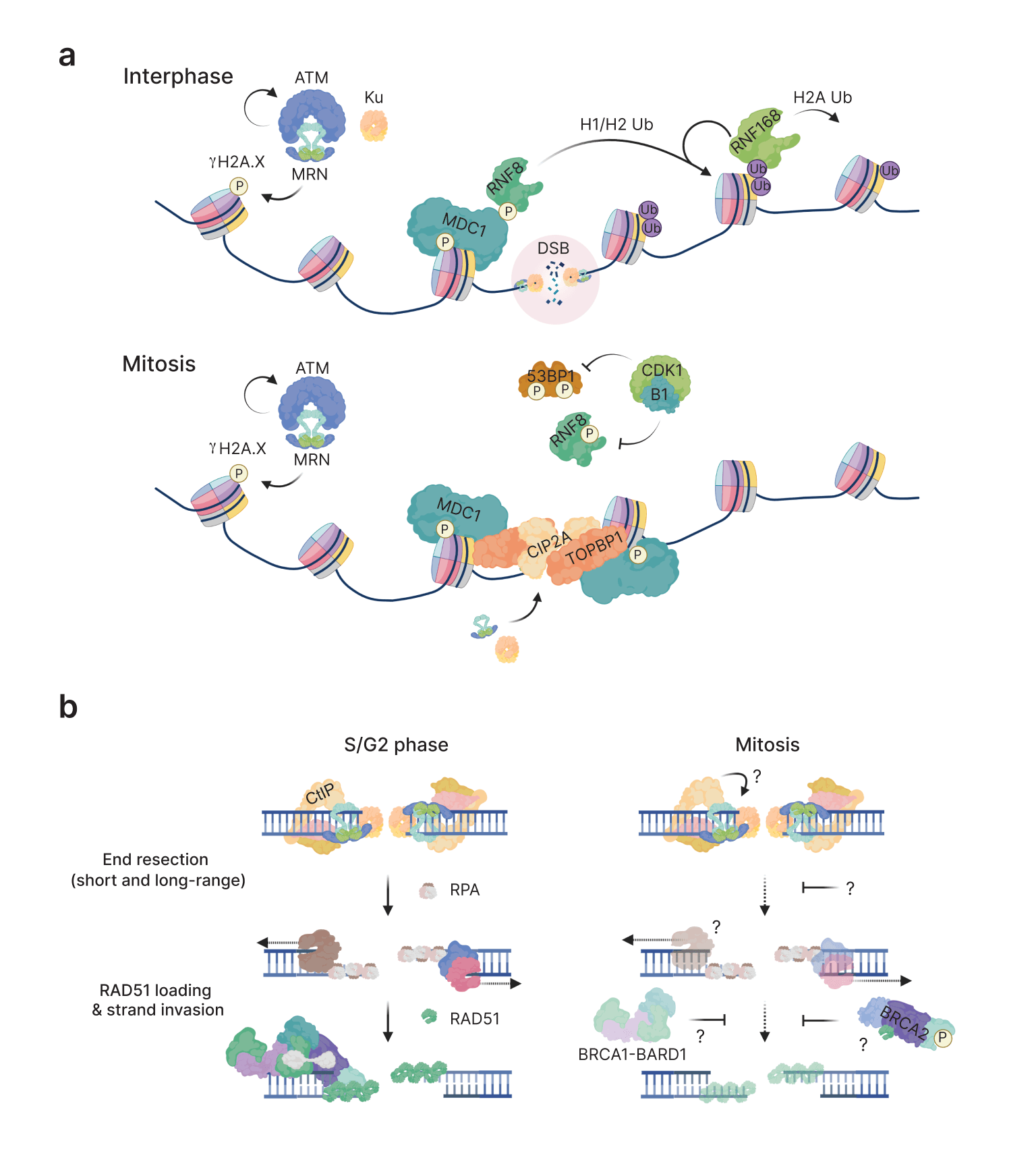
Schematic highlighting differences between interphase and mitotic DSB responses. Bottom: HR is minimally inhibited in mitosis by targeting of both end resection and strand invasion mechanisms.
Characterization of the MDC1-CIP2A-TOPBP1 mitotic DNA damage tolerance complex
My PhD lab and collaborators from Manuel Stucki's lab in Zurich found that MDC1 interacts with TOPBP1 and CIP2A specifically in mitotic cells. CIP2A responds to both DNA replication associated lesions and exogenous lesions, such as those induced by X-ray IR. My lab and I found that CIP2A and TOPBP1 are recruited to mitotic DSBs in a co-dependent manner independent of the PP2A phosphatase regulatory function of CIP2A. Moreover, Salomé Adam, Silvia Rossi, and Nicole Hustedt in my lab found that CIP2A loss is synthetic lethal with BRCA1 and BRCA2 loss, implicating CIP2A as a selective target for BRCA-mutated cancers. This work was published in Nature Cancer in 2021. Ongoing work is under way to test the therapeutic benefit of targeting the CIP2A-TOPBP1 interaction in BRCA-mutated settings.
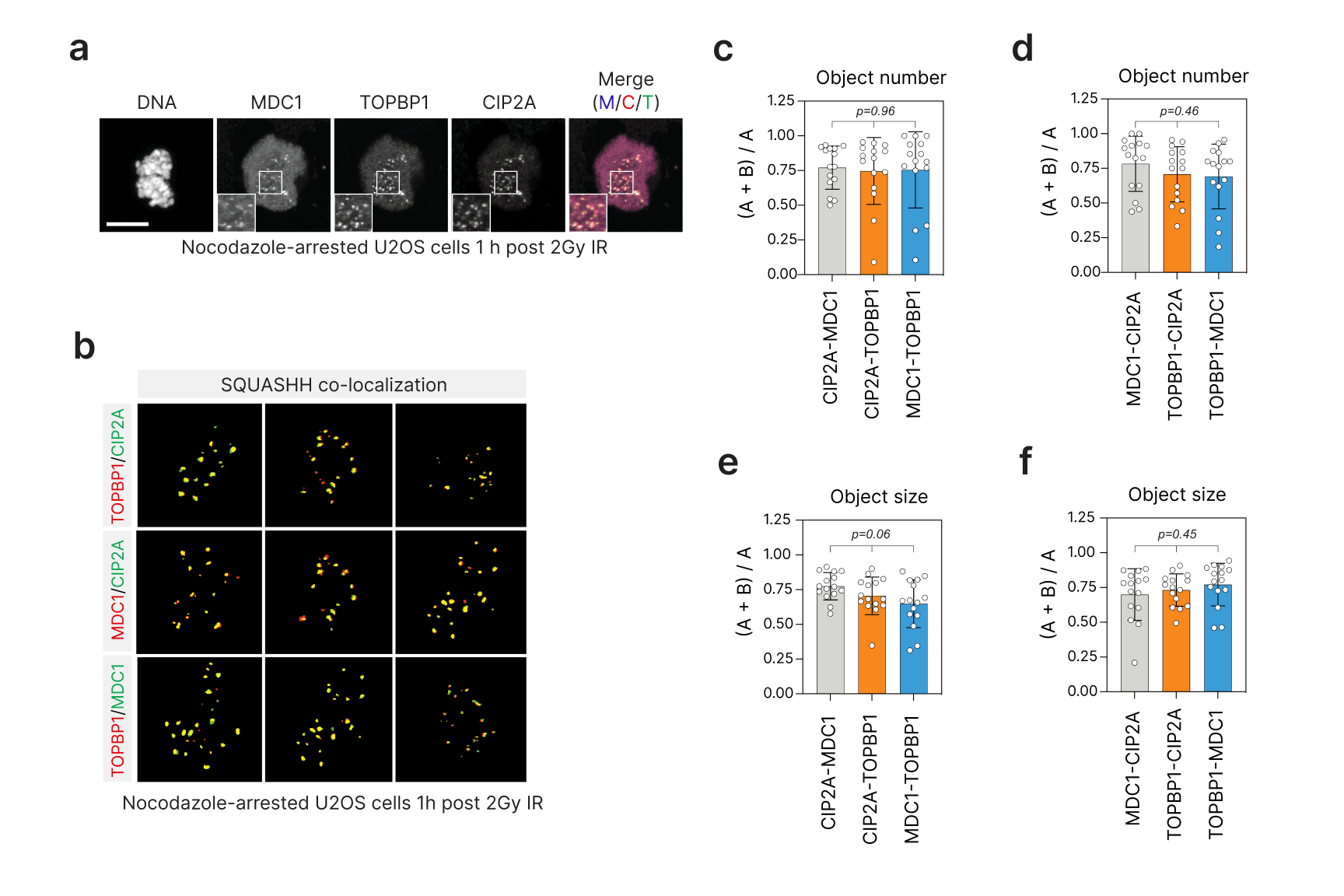
Immunofluorescence of the MDC-TOPBP1-CIP2A complex in response to X-ray irradiation. Co-localization was quantified using the Segmentation and Quantification of Subcellular Structures (SQUASHH) algorithm. Values greater than 0.5 indicate co-localization. Statistical analysis by one-way ANOVA.
Design Outcome
My work harnessed the power of forward genetic approaches to answer biological questions. I found an unexpected activation of the stress-responsive kinase GCN2 that drives the cytotoxic effects of adavosertib and other WEE1 kinase inhibitors. Moreover, I found several interesting genetic interactions with mirin and its chemical derivatives. I also uncover important mechanisms of DNA damage regulation in mitotic cells. In particular, targeting of the MDC1-TOPBP1-CIP2A complex may prove to be useful for a subset of BRCA-mutated cancers. Want to read more? Connect with me for a copy of my thesis.